Calibrating species_radii
species_radii is the element-wise dictionary that controls how the free-volume Monte Carlo estimator decides whether a sampled point is occupied. Because \(V_{\mathrm{free}}\) enters the GCMC insertion and deletion acceptance terms directly (see Free volume and species_radii), the quality of these radii is a sensitive input – not a tuning knob to leave at default values.
The exclusion radius for a host species should reflect the shortest adsorbate-host distance that survives a local relaxation with the calculator used in production (the thesis behind this library denotes it \(r_{\mathrm{relax}}\)). Every value of species_radii is tied to the specific calculator and relaxation settings used during production; re-calibrate if either changes.
Free-volume estimate (recap)
For completeness, the estimator used by every cell object in mcpy is:
See Free volume and species_radii for the role of \(V_{\mathrm{free}}\) in the GCMC acceptance terms.
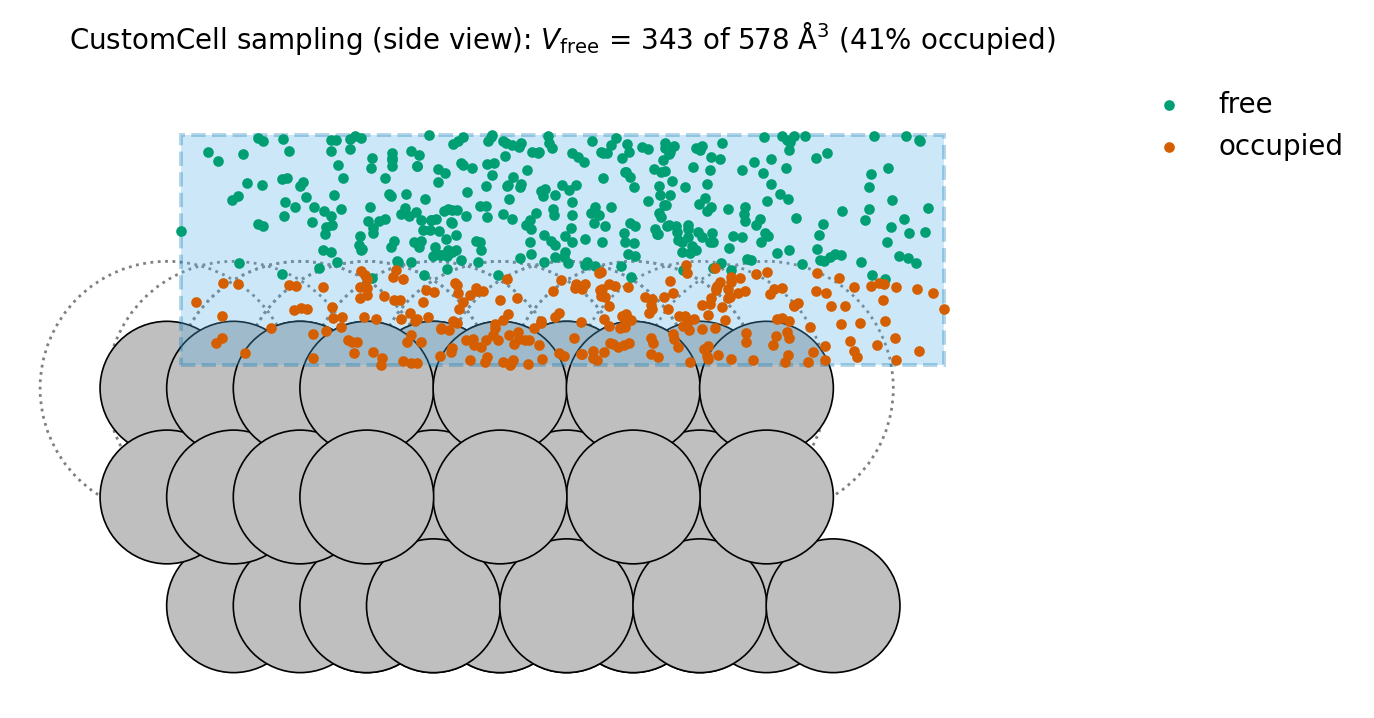
The estimator at work in a CustomCell over an Ag(111) slab, sampled
by the cell itself (docs/make_figures.py). Each point drawn by
cell.get_random_point() is classified against the exclusion disks of
radius \(r_{\mathrm{Ag}}\) (dotted circles); the free fraction sets
the \(V_{\mathrm{free}}\) that cell.get_volume() returns.
Measure the radius from relaxed insertions
Drop a trial adsorbate at random positions above the surface, relax each trial with your production calculator, and record the nearest adsorbate-host distance that survives:
import numpy as np
from ase.build import fcc111
from ase.constraints import FixAtoms
from ase.optimize import LBFGS
rng = np.random.default_rng(11)
slab0 = fcc111('Ag', a=4.085, size=(3, 3, 3), vacuum=8.0, periodic=True)
z_top = slab0.positions[:, 2].max()
distances = []
for _ in range(150):
slab = slab0.copy()
slab.set_constraint(FixAtoms(indices=[a.index for a in slab if a.tag == 3]))
slab.append('O')
slab.positions[-1] = [rng.uniform(0, slab.cell[0, 0]),
rng.uniform(0, slab.cell[1, 1]),
z_top + rng.uniform(1.0, 3.5)]
slab.calc = calculator # your production calculator
LBFGS(slab, logfile=None).run(fmax=0.1, steps=50)
d = slab.get_distances(len(slab) - 1, range(len(slab) - 1), mic=True)
distances.append(d.min())
r_relax = np.percentile(distances, 5) # edge of the first peak
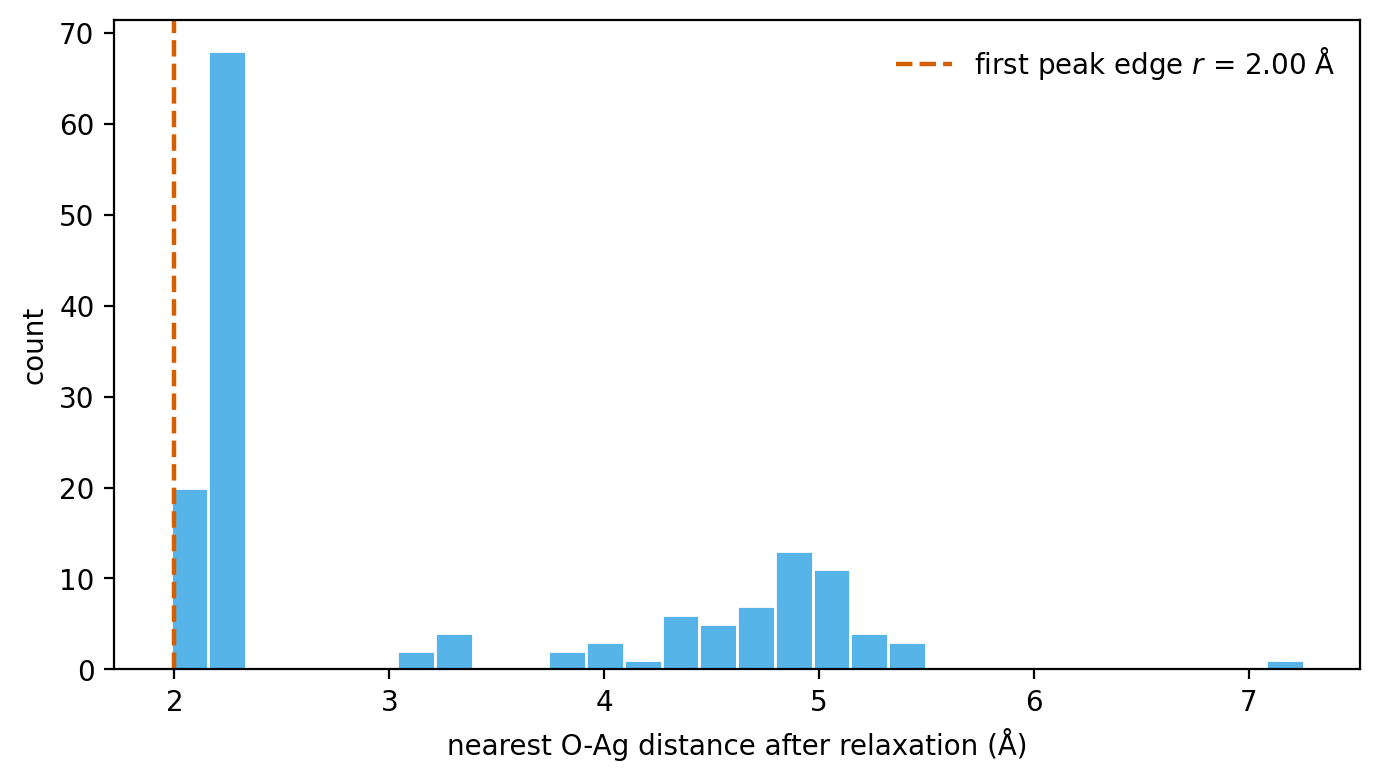
The histogram this loop produces (O on Ag(111); EMT stands in for the
production calculator, generated by docs/make_figures.py). The sharp
first peak collects the chemisorbed outcomes; its lower edge is
\(r_{\mathrm{relax}}\).
The far cluster of large distances comes from trials that relaxed away without binding –
ignore it and read off the lower edge of the first peak.
scripts/compute_radii.py packages this loop for FCC(111) hosts with a MACE model.
Use the value in a simulation
Put the measured pair distance on the host species and zero on the adsorbate (or split it equally between the two – either convention works if used consistently):
cell = CustomCell(
atoms,
custom_height=5.0,
bottom_z=atoms.positions[:, 2].max() + 0.5,
species_radii={'Ag': r_relax, 'O': 0.0},
)
Validate with a short pilot run before production: the insertion acceptance ratio and the reported \(V_{\mathrm{free}}\) should both be stable and non-degenerate.